

En los campos de la biogeografía y la biología evolutiva, investigar la distribución de las especies y su relación con la evolución es esencial para desentrañar los complejos patrones que han dado forma a la diversidad de la vida a lo largo del tiempo y en diferentes lugares.

Contenido del post
Introducción
En los campos de la biología evolutiva y la biogeografía, investigar la distribución de las especies y su relación con la evolución es esencial para desentrañar los complejos patrones que han dado forma a la diversidad de la vida a lo largo del tiempo y en diferentes lugares. La distribución geográfica de las especies es el resultado de una interacción dinámica entre una multitud de factores. Entre estos factores se incluyen eventos históricos, como las glaciaciones que han modificado los paisajes y los hábitats; cambios climáticos a lo largo de escalas temporales diversas, que han influido en la disponibilidad de recursos y en la adaptabilidad de las especies; barreras geográficas, como cordilleras, ríos y océanos, que han actuado como obstáculos para la dispersión; y procesos evolutivos, como la evolución de características morfológicas y fisiológicas que han permitido a las especies adaptarse a entornos específicos y expandirse a nuevas áreas.
En este tutorial, exploraremos cómo utilizar la función

Código en R
Para poder realizar este análisis vamos a utilizar el código que encontraremos en phylo.to.map para análisis biogeográficos, el cual es un repositorio creado por el autor, con el fin de compartir su aprendizaje. En este repositorio encontrarás los archivos relevantes para el análisis. Recuerda que es esencial asegurar que R pueda acceder a los archivos necesarios sin problemas.
El primer paso que daremos será cargar los paquetes necesarios que utilizaremos en este tutorial mediante la función
library(phytools)
library(viridis)
library(viridisLite)
library(ape)
library(maps)Despues de cargados los paquetes necesarios, procederemos a leer los archivos requeridos para nuestro análisis, para ello, utilizaremos las funciones

Los datos que cargaremos están ordenados en una matriz que contiene la latitud y la longitud de todas las especies a analizar.
Debes tener en cuenta que los nombres de las filas deben ser los mismos que los de tree$tip.label; sin embargo, se puede proporcionar más de un conjunto de coordenadas por especie duplicando algunos nombres de fila.
data <- read.csv("https://raw.githubusercontent.com/Maxornatus/phylo.tomap/main/coo_vari.csv",
header = TRUE, row.names = 1)
tree <- read.tree("https://raw.githubusercontent.com/Maxornatus/phylo.tomap/main/B_vari.tre")Posteriormente a nuestra lectura de datos, tendremos que definir una lista de rangos de filas que corresponden a diferentes grupos de datos, esta lista está previamente definida según los análisis filogenéticos previos. Estos rangos se utilizarán posteriormente para asignar colores a cada grupo en las visualizaciones.
rangos_color <- list(c(1, 12), # Primer rango de la especie 1 a la 12
c(13, nrow(data))) # Segundo rango de la especie 13 en adelante.Luego, estableceremos un vector de colores personalizados que se asignarán a cada uno de los dos grupos de datos. Estos colores se utilizarán para distinguir visualmente los diferentes grupos. En nuestro caso, utilizaremos el color azul y verde.
colores_personalizados <- c("blue", "green")Función phylo.to.map()
Ahora utilizaremos la función

obj <- phylo.to.map(tree, data, plot = FALSE, direction = "rightwards")Luego, inicializaremos un vector para almacenar los colores asignados a cada especie dentro de los grupos definidos anteriormente. Este paso se realiza con el fin de preparar el terreno para asignar colores específicos a cada especie.
cols <- rep(NA, length(obj[["tree"]][["tip.label"]]))Posteriormente, realizaremos un bucle, esto con el fin de asignar colores a cada especie de nuestro estudio dentro de los grupos definidos anteriormente. Así, los colores se asignan de acuerdo con los rangos de filas establecidos previamente.
for (i in seq_along(rangos_color)) {
rango <- rangos_color[[i]]
colores_rango <- rep(colores_personalizados[i], diff(rango) + 1)
cols[rango[1]:rango[2]] <- colores_rango
}Ahora, nos aseguraremos que todas las especies reciban un color, incluso aquellas que no se hayan asignado explícitamente. Esto garantiza una representación visual completa en las visualizaciones posteriores.
cols[is.na(cols)] <- "gray"Sumado a ello, vamos a asignar colores a las puntas de nuestro árbol filogenético, lo que permite visualizar la relación entre las poblaciones y su distribución geográfica.
cols <- setNames(cols, tree$tip.label)Visualización y análisis
Para generar una visualización mediante un mapa filogenético, utilizaremos la función

plot(obj, direction = "rightwards", colors = cols, ftype = "off", fsize = 0.8,
cex.points=c(0.8,1.2), pts = T, lwd = 1.5, xlim = c(-90, -35), ylim = c(-55, 12),)
legend("topright", legend = c("Población uno", "Población dos"),
fill = colores_personalizados, cex = 0.8, bty = "n")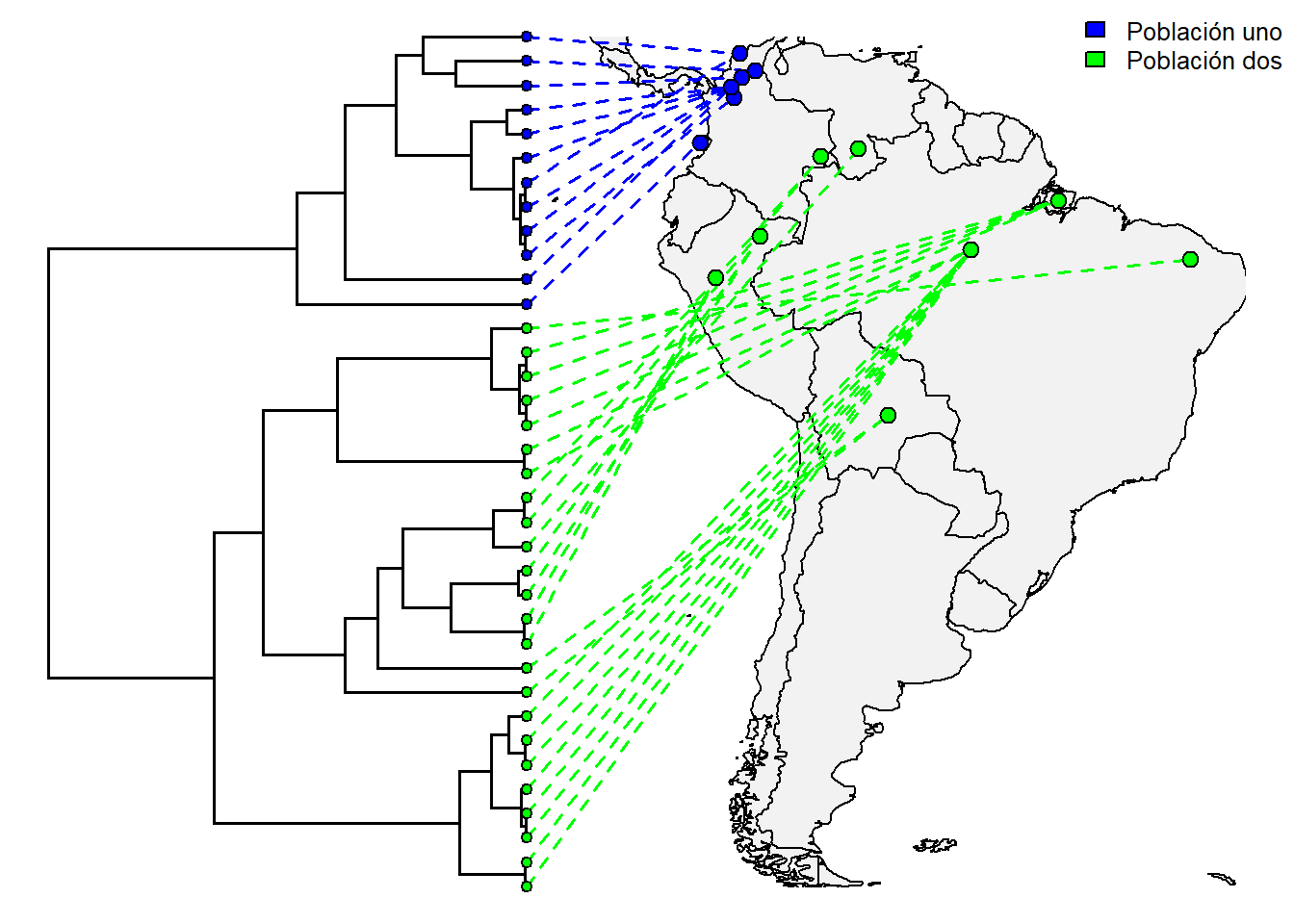
Cómo podemos ver, ya nuestro mapa está generado, incluyendo el árbol filogenético de las especies y su distribución en sudamérica. Además, podremos incluir los nombres de los individuos en las puntas del árbol
plot(obj, direction = "rightwards",colors = cols, ftype = "i", fsize = 0.8,
cex.points = c(0.8, 1.2), pts = T, lwd = 1.5, xlim = c(-90, -35),
ylim = c(-55, 12),)
# Personalizamos la leyenda de nuestro mapa
legend("topright", legend = c("Poblacion uno", "Poblacion dos"),
fill = colores_personalizados, cex = 0.8, bty = "n")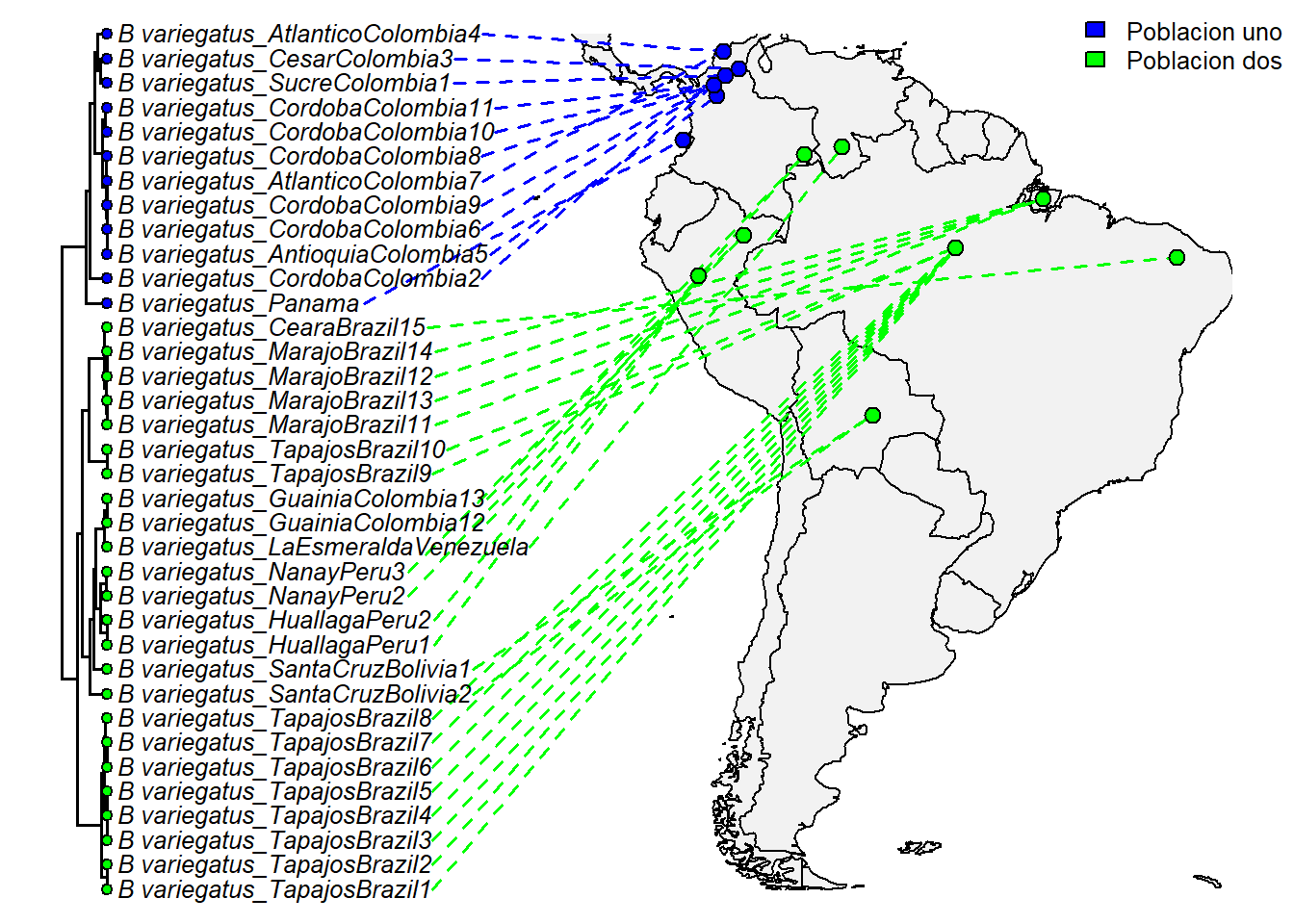
Así, nuestro mapa está completado con las relaciones de las diferentes especies analizadas y su relación en relación a la distribución biogeográfica de cada especie. Igualmente, podemos generar una visualización alternativa que muestre la relación filogenética dentro del mapa geográfico, proporcionando una perspectiva adicional sobre la distribución y la evolución de las poblaciones.
plot(obj, type = "direct", colors = cols, pts = FALSE, fsize = 0.8,
xlim = c(-90, -35), ylim = c(-55, 12), map.bg = "lightgreen",
map.fill = "lightblue", ftype = "off", cex.points = 3, delimit_map = TRUE)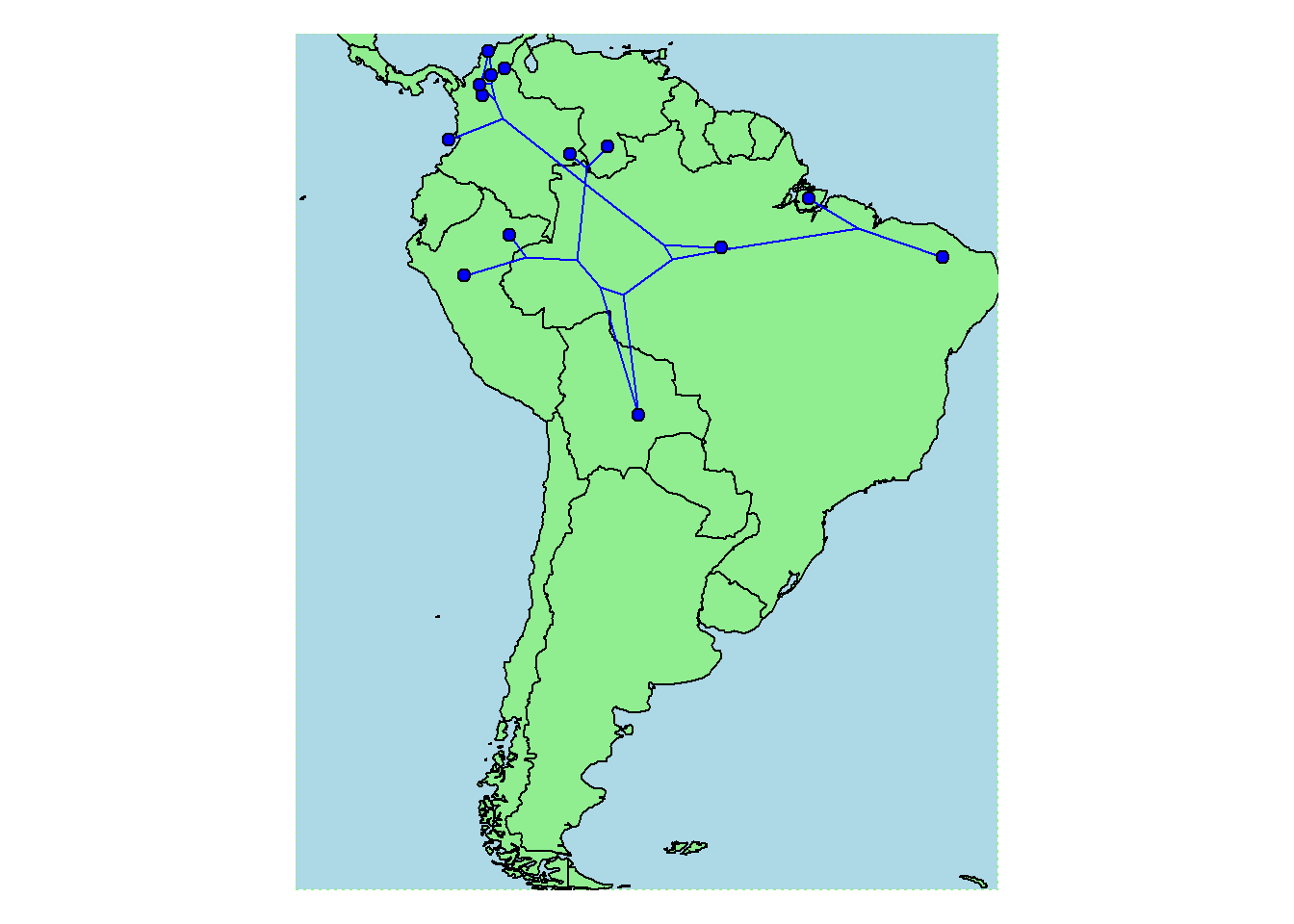
Resumen
Cada paso en este código contribuye a la preparación y visualización de nuestros datos para comprender mejor la distribución geográfica y la relación evolutiva de las poblaciones o especies que estamos estudiando.
Recuerda que debemos hacer un análisis reflexivo sobre nuestros resultados para comprenderlos de mejor manera y poder desarrollar una discusión y conclusión más acertada de nuestra investigación.

Literatura
Más información
Estos análisis se han realizado utilizando el software R (v.4.3.2) y Rstudio (v. 2023.12.1+402).
El proceso de edición del post ha sido realizado por David Vanegas-Alarcón
Recuerda que todos nuestros códigos están almacenados en GitHub
